Cette section vous permet de consulter nos dernières actualités AFAF sur la recherche. Vous pouvez les retrouver aussi avec nos actualités générales sur la page Actualités.
L’AFAF vous propose une nouvelle session d’explication de texte scientifique (ou lecture critique d’article) à partir de l’article suivant :
Peripheral frataxin levels govern long-term clinical progression in Friedreich ataxia, publié en mars 2026 dans BMJ Neurology Open, et disponible en ligne.
Vous trouverez ci-dessous les dernières informations communiquées par le groupe Biogen.
Étude de phase 3 BRAVE (pédiatrie) – Une étape atteinte en matière d’inclusion
L’étude BRAVE a atteint 50% de son objectif d’inclusion, avec 128 participants recrutés à ce jour. Pour rappel, l’étude prévoit d’inclure jusqu’à 255 participants.
L’étude inclut désormais des enfants âgés de 2 à moins de 7 ans, en plus de ceux âgés de 7 à 15 ans. Cette étape importante nous permet de tenter de répondre aux besoins non couverts d’une population pédiatrique plus large, quels que soient l’âge et la capacité de marche des patients (ambulatoires et non ambulatoires).
Pour rappel, l’étude BRAVE est un essai clinique international de phase 3 évaluant l’efficacité et la sécurité de l’omaveloxolone chez des enfants atteints d’ataxie de Friedreich et âgés de 2 à 15 ans. Le recrutement pour l’étude BRAVE a débuté au mois de mai 2025 et se poursuit actuellement dans plusieurs centres cliniques à travers le monde.
Les centres actuellement actifs en France sont les suivants :
• Paris : AP-HP – Hôpital Armand Trousseau
• Montpellier : CHU de Montpellier – Hôpital Gui de Chauliac
Les informations les plus récentes concernant l’étude de phase 3 BRAVE sont publiées sur clinicaltrials.gov (NCT06953583). Les personnes intéressées par cette étude sont invitées à en discuter avec leur professionnel de santé.
Pour rappel, l’omaveloxolone n’est à ce jour pas autorisé dans le traitement des patients âgés de 2 à 15 ans. L’omaveloxolone est indiqué dans le traitement de l’ataxie de Friedreich chez les personnes âgées de 16 ans et plus.
Fin de l’étude d’extension en ouvert (OLE) MOXIe
Le dernier participant a désormais terminé l’étude d’extension en ouvert (OLE) MOXIe.
L’étude MOXIe était un essai international multicentrique, randomisé, en double aveugle et contrôlé par placebo, suivi d’une phase d’extension en ouvert. Elle visait à mieux comprendre la sécurité de l’omaveloxolone ainsi que son impact sur l’effort physique, le mouvement, la coordination et la qualité de vie des personnes atteintes vivant avec l’ataxie de Friedreich.
L’étude MOXIe a joué un rôle clé dans les autorisations réglementaires de l’omaveloxolone pour le traitement de l’ataxie de Friedreich chez les patients âgés de 16 ans et plus. La phase d’extension en ouvert a permis d’évaluer la sécurité et l’efficacité du traitement sur une durée allant jusqu’à trois ans.
La finalisation de cette étude constitue une étape importante rendue possible grâce à l’implication des participants, de leurs familles, des investigateurs, des équipes des centres et de l’ensemble de la communauté de l’ataxie de Friedreich.
Nous espérons partager prochainement les données finales de l’étude MOXIe lors d’un congrès scientifique.
Exploration d’une forme de comprimé pour suspension buvable comme mode d’administration alternatif de l’omaveloxolone
Biogen a lancé récemment une étude de phase 1 visant à évaluer la bioéquivalence d’une forme « comprimé » d’omaveloxolone, destinée à être administrée sous forme de suspension buvable, comparativement à la gélule orale actuellement disponible.
À ce jour, l’omaveloxolone est administré par voie orale sous forme de gélules, ou avec le contenu des gélules saupoudré sur de la compote de pommes. L’objectif principal de cette étude est d’évaluer si l’omaveloxolone est absorbé et métabolisé de manière comparable chez des adultes en bonne santé lorsqu’il est administré sous forme de suspension buvable par rapport aux gélules.
Biogen est conscient que certaines personnes peuvent rencontrer des difficultés à avaler des gélules et souhaite explorer des options supplémentaires d’administration de l’omaveloxolone. Si cette étude est concluante et que cette nouvelle formulation est approuvée par les autorités réglementaires, elle pourrait permettre une administration du traitement sous forme liquide. Les informations les plus récentes concernant cette étude sont et seront publiées sur ClinicalTrials.gov (NCT07297199).
Le 20 mars 2026, le Dr Russell Clayton, directeur médical de Larimar Therapeutics, a présenté les dernières informations concernant le programme clinique du nomlabofusp pour l’ataxie de Friedreich.
La partie “Questions-Réponses” du webinaire ne fait pas partie de cette rediffusion.
L’AFAF vous propose une nouvelle session d’explication de texte scientifique (ou lecture critique d’article) à partir de l’article suivant : Alpha-lipoic acid supplementation improves pathological alterations in cellular models of Friedreich ataxia (La supplémentation en acide alpha-lipoïque améliore les altérations pathologiques dans les modèles cellulaires de l’ataxie de Friedreich), publié en août 2025 et disponible en ligne.
Nous vous enverrons par la suite un lien de connexion.
N’hésitez pas à contacter le groupe recherche pour toute question à recherche@afaf.asso.fr.
Publications récentes
Nous relayons ici des publications scientifiques récentes ayant trait à l’ataxie de Friedreich et qui ne seraient pas détaillées dans la section ‘nos dernières informations‘.
Vous trouverez la traduction du titre de l’article, les références et le lien vers la publication correspondante.
Vous pouvez contacter les membres du groupe recherche AFAF pour tout complément sur les articles mentionnés, par l’adresse recherche@afaf.asso.fr.
Cette section n’est pas exhaustive. Les articles ou publications qui font l’objet de traductions par l’AFAF, ou par d’autres associations, sont disponibles sur les autres pages de notre site, vous en serez informés dans la section.
Retrouvez par ailleurs notre bibliographie d’articles scientifiques sur la page de veille bibliographique.
Christian Rummey, Ian A Blair, Clementina Mesaros, Teerapat Rojsajjakul, Yina Dong, George Wilmot, Theresa Zesiewicz, Kathy Mathews, Joseph C Hoyle, Lauren Seeberger, Louise A Corben, Martin Bruce Delatycki, Richard S Finkel, Richard H Roxburgh, Antoine Duquette, Grace Yoon, Christopher M Gomez, S H Subramony, Susan Perlman, Shana Mccormack, David R Lynch
Dans cet article publié en mars 2026, les auteurs observent que des taux plus bas de frataxine dans le sang (avec deux méthodes différentes de quantification) sont associés à un début plus précoce des symptômes, à une perte plus rapide de la marche et à une progression clinique plus importante. L’intérêt principal est que la mesure de la frataxine dans le sang pourrait aider à suivre l’évolution de la maladie et à évaluer l’effet de futurs traitements visant à augmenter cette protéine. Les auteurs concluent que la frataxine périphérique est un biomarqueur clinique pertinent, avec un intérêt potentiel pour les essais thérapeutiques.
Cet article a fait l’objet d’une « Lecture critique d’article » le 12 mai 2026, visionnable sur le site AFAF.
Résumé de l’article traduit par l’équipe recherche.
(Des notes en bas de page visent à aider la compréhension)
Contexte Les nouvelles thérapeutiques de l’ataxie de Friedreich utilisent diverses stratégies visant à augmenter les taux de protéine frataxine. Une meilleure compréhension du lien entre ces taux et les résultats cliniques pourrait renforcer leur utilisation comme marqueurs pharmacodynamiques, et potentiellement comme critère de substitution dans le développement thérapeutique. Un cadre de modélisation élaboré a été développé afin d’évaluer la pertinence de la frataxine comme biomarqueur selon les méthodes de dosage, les tissus et les stades de la maladie.
Méthodes Les taux de frataxine avaient été générés antérieurement à l’aide de deux plateformes analytiques distinctes et à partir de deux cohortes cliniques séparées : la frataxine dans le sang total a été mesurée par immunodosage à flux latéral [1] (cohorte LF[2]), et par une méthode LC-MS/MS à triple quadripôle[3] (cohorte TQ[4]), qui permet la quantification séparée de la frataxine mature (FXN-M)[5] et de la frataxine spécifique des érythrocytes (FXN-E)[6]. Les résultats ont été comparés de façon descriptive à ceux de témoins et de porteurs hétérozygotes, et plusieurs stratégies de modélisation distinctes ont été utilisées pour les corréler à la fonction clinique.
Résultats Les deux cohortes représentaient le spectre pertinent de la maladie, avec des différences mineures de sévérité génétique et clinique, toutes deux corrélées aux taux de frataxine[7]. Les porteurs hétérozygotes présentaient des taux intermédiaires. La modélisation a confirmé la valeur prédictive de la frataxine à travers plusieurs évaluations cliniques, telles que l’âge de début des symptômes, l’âge de perte de la marche et la progression à long terme. GAA1, l’expansion de répétition la plus courte, a été confirmé comme le principal prédicteur de la frataxine elle-même et, dans la plupart des situations, de la fonction clinique.
Discussion et conclusion Bien que la biologie des isoformes[8] et l’expression spécifique aux tissus[9] demeurent des éléments importants à considérer, la quantification périphérique de la frataxine fournit une mesure biologiquement fondée de la physiopathologie et de la progression de la maladie, avec un fort potentiel d’application dans les essais thérapeutiques. La frataxine est un biomarqueur clinique valide, et nos résultats soutiennent la poursuite de son développement comme critère de substitution dans l’ataxie de Friedreich.
[1] Un immunodosage à flux latéral est un test biologique qui fonctionne un peu comme un test rapide — par exemple certains tests de grossesse ou tests antigéniques. On dépose un échantillon, puis celui-ci migre sur une bandelette où des anticorps reconnaissent spécifiquement la molécule recherchée. Ici, le test sert à mesurer la frataxine, la protéine diminuée dans l’ataxie de Friedreich.
[2] La cohorte LF désigne donc le groupe de patients chez qui la frataxine a été mesurée avec cette méthode de type lateral flow.
[3] La LC-MS/MS signifie chromatographie liquide couplée à la spectrométrie de masse en tandem. C’est une méthode de laboratoire très précise qui permet d’identifier et de mesurer de très petites quantités de molécules dans un échantillon biologique.
[4] La cohorte TQ correspond donc au groupe de patients chez qui la frataxine a été mesurée par cette méthode LC-MS/MS, plus technique et très précise.
[5] La quantification séparée de FXN-M et FXN-E signifie que le laboratoire ne mesure pas seulement une quantité globale de frataxine dans le sang : il distingue deux formes différentes de cette protéine. FXN-M, ou frataxine mature, correspond à la forme principale de la frataxine, présente dans la plupart des cellules et surtout liée aux mitochondries, les petites structures qui produisent l’énergie de la cellule. Cette forme est importante car la frataxine participe notamment au bon fonctionnement des protéines contenant des centres fer-soufre.
[6]FXN-E, ou frataxine spécifique des érythrocytes, correspond à une autre forme de frataxine retrouvée surtout dans les globules rouges. Elle est particulière parce que les globules rouges matures n’ont pas de mitochondries ; FXN-E n’est donc pas exactement l’équivalent de la frataxine mitochondriale classique. De façon simplifiée: la méthode LC-MS/MS à triple quadripôle permet de dire séparément combien il y a de frataxine “classique” FXN-M et combien il y a de frataxine liée aux globules rouges FXN-E. C’est utile car, dans un prélèvement sanguin, les globules rouges sont très nombreux : si l’on ne distingue pas les deux formes, on risque de mélanger des informations biologiques différentes. L’article indique précisément que la cohorte TQ utilise cette méthode parce qu’elle permet cette quantification séparée de FXN-M et FXN-E.
[7] Après exclusion de certains participants, les chercheurs ont étudié 391 patients dans une première cohorte et 245 dans une seconde. Les deux groupes n’étaient pas exactement comparables : l’un comportait davantage de formes à début plus tardif, tandis que l’autre regroupait plus de formes précoces ou typiques. Malgré ces différences, les deux cohortes représentaient bien l’ensemble des formes de sévérité de la maladie, ce qui permet d’étudier de façon fiable l’évolution clinique à long terme.
[8] La biologie des isoformes désigne le fait qu’un même gène peut donner naissance à plusieurs versions légèrement différentes d’une protéine. On peut l’imaginer comme une même recette de base qui, selon les étapes utilisées, produit plusieurs variantes du même plat. Ces variantes sont appelées isoformes. Elles peuvent différer par leur longueur, leur composition, leur localisation dans la cellule ou parfois leur fonction. Dans le cas de la frataxine, cela signifie que l’on ne parle pas forcément d’une seule forme uniforme de la protéine. La frataxine mature, ou FXN-M, correspond à la forme mitochondriale, présente dans les cellules contenant des mitochondries. La frataxine E, ou FXN-E, est une isoforme extramitochondriale retrouvée surtout dans les érythrocytes, c’est-à-dire les globules rouges. L’intérêt de distinguer les isoformes est donc très concret : si l’on mesure seulement la quantité totale de frataxine dans le sang, on peut mélanger des informations biologiques différentes. Mesurer séparément FXN-M et FXN-E permet de savoir plus précisément quelle forme de frataxine est évaluée, dans quel type de cellule, et avec quelle signification biologique potentielle. Cela ne veut pas dire automatiquement que les deux isoformes ont la même fonction ou la même valeur clinique.
[9] L’expression « spécifique aux tissus » signifie qu’une molécule, un gène, une protéine ou une isoforme est présente, produite ou active surtout dans certains tissus de l’organisme, et pas de manière identique partout. Un tissu est un ensemble de cellules spécialisées : par exemple le tissu musculaire, le tissu nerveux, le foie, le sang, la peau, etc. Toutes les cellules possèdent globalement le même ADN, mais elles n’utilisent pas les mêmes gènes de la même façon. Ainsi, une cellule du foie ne fabrique pas exactement les mêmes protéines qu’un neurone ou qu’un globule rouge. Dire qu’une protéine est spécifique d’un tissu ne veut pas toujours dire qu’elle existe uniquement dans ce tissu. Cela peut signifier qu’elle y est beaucoup plus abondante, qu’elle y joue un rôle particulier, ou qu’une forme particulière de cette protéine y est surtout retrouvée.
Cette revue Cochrane actualisée, publiée en mai 2026, les auteurs ont analysé plusieurs essais cliniques évaluant des médicaments dans l’ataxie de Friedreich.
Résumé de l’article traduit par l’équipe recherche
(Des notes en bas de page visent à aider la compréhension)
Justification : L’ataxie de Friedreich est une maladie neurologique héréditaire rare, de transmission autosomique récessive. Elle se caractérise d’abord par une instabilité en station debout et à la marche, puis évolue lentement vers une dépendance au fauteuil roulant, généralement à la fin de l’adolescence ou au début de la vingtaine. Une scoliose, des pieds creux et une cardiomyopathie sont souvent présents au moment du diagnostic. À mesure que la maladie progresse, les personnes atteintes développent habituellement une parole dysarthrique, des troubles auditifs — surtout en environnement bruyant —, des troubles urinaires, un diabète, de l’anxiété, une dépression, une spasticité musculaire et des troubles visuels. Les anomalies cardiaques sont responsables d’un décès prématuré chez 60 % des personnes atteintes d’ataxie de Friedreich. Il n’existe pas de marqueur clinique ou biologique simple permettant d’évaluer la progression de la maladie, ni de traitement curatif connu. Il s’agit de la troisième mise à jour d’une revue publiée en 2009, puis actualisée en 2012 et en 2016.
Objectifs :
Évaluer les effets des traitements pharmacologiques chez les personnes atteintes d’ataxie de Friedreich après 12 mois de traitement.
Méthodes de recherche :
Pour identifier les études à inclure dans cette revue, les auteurs ont recherché les essais dans CENTRAL, MEDLINE, Embase, CINAHL Plus, TRIP, Orphanet, la plateforme internationale d’enregistrement des essais cliniques de l’OMS — ICTRP — et ClinicalTrials.gov. Ils ont également vérifié les listes de références des études pertinentes et contacté les auteurs des études. La recherche la plus récente a été effectuée le 4 février 2025.
Critères d’éligibilité :
Les auteurs se sont intéressés aux essais contrôlés randomisés[1] et quasi randomisés portant sur des traitements pharmacologiques, y compris les vitamines, chez des personnes ayant une ataxie de Friedreich génétiquement confirmée. Pour être incluses dans la revue, les études devaient durer au moins 12 mois.
Critères de jugement :
Les auteurs ont évalué, après 12 mois de traitement, les critères suivants : variation du score sur une échelle validée d’évaluation de l’ataxie ; variation de l’épaisseur du septum interventriculaire en diastole[2], mesurée par IRM cardiaque ou échocardiographie ; variation des activités de la vie quotidienne à l’aide d’un questionnaire validé ; variation de la dextérité des membres supérieurs ; et variation des résultats aux tests d’effort cardio-pulmonaire. Ils ont également évalué les effets indésirables liés au traitement lorsque le médicament était poursuivi pendant toute la durée de l’étude, ainsi que les effets indésirables apparus sous traitement ayant conduit à l’arrêt du médicament ou au décès pendant la période d’étude.
Risque de biais :
À l’aide de l’outil Cochrane RoB 2 d’évaluation du risque de biais, les auteurs ont évalué le risque de biais pour les sept critères de jugement présentés dans leurs tableaux récapitulatifs des résultats.
Méthodes de synthèse :
Les résultats des études ont été synthétisés pour chaque critère de jugement au moyen d’une méta-analyse[3] lorsque cela était possible. Les auteurs ont calculé la différence moyenne ou la différence moyenne standardisée[4] pour les critères continus[5], et le risque relatif pour les critères dichotomiques[6], avec des intervalles de confiance à 95 %[7]. Ils ont utilisé la méthode GRADE[8] pour évaluer le niveau de certitude des données concernant les critères de jugement prédéfinis, classé comme élevé, modéré, faible ou très faible.
Études incluses :
Huit essais contrôlés randomisés ont été inclus dans cette revue mise à jour. Sept d’entre eux ont été inclus dans la méta-analyse ; ces études regroupaient au total 574 participants, avec des effectifs allant de 29 à 232 participants par étude. Une étude était internationale, avec des centres en Amérique du Nord, en Europe et en Australie. Quatre autres études étaient multicentriques : trois en Europe et une en Amérique du Nord. Les études monocentriques ont été menées au Royaume-Uni, en Italie et en France. Les participants avaient entre 8 et 70 ans au moment de l’inclusion, avec un âge moyen compris, selon les études, entre 18 et 35 ans ; l’âge moyen global non pondéré était de 25,9 ans. Toutes les études incluaient des hommes et des femmes, la proportion de femmes variant de 21 % à 58 %. Soixante-huit pour cent des participants présentaient une ataxie sévère, tandis que 32 % avaient une ataxie modérée. Les traitements étudiés étaient l’érythropoétine alpha, la CoQ10 associée à la vitamine E, l’idébénone, le lériglitazone, l’omaveloxolone et le RT001. Une étude a testé la pioglitazone, mais n’a publié aucun résultat. Quatre études étaient contrôlées par l’industrie pharmaceutique.
Synthèse des résultats :
La méta-analyse de sept études a montré que les traitements pharmacologiques font probablement peu ou pas de différence sur les scores d’évaluation de l’ataxie après 12 mois de traitement, par rapport au placebo : différence moyenne standardisée 0,02 ; intervalle de confiance à 95 % de -0,23 à 0,26 ; I² = 42 % ; 7 études ; 513 participants ; niveau de certitude modéré.
Les données étaient très incertaines concernant les effets du traitement sur l’épaisseur du septum interventriculaire en diastole : différence moyenne -0,51 ; intervalle de confiance à 95 % de -1,10 à 0,09 ; I² = 80 % ; 2 études ; 72 participants ; niveau de certitude très faible. Elles étaient également très incertaines pour les activités de la vie quotidienne : différence moyenne -0,59 ; intervalle de confiance à 95 % de -1,39 à 0,21 ; I² = 24 % ; 3 études ; 167 participants ; niveau de certitude très faible.
La méta-analyse de trois études a montré que le traitement améliore probablement la dextérité des membres supérieurs : différence moyenne standardisée -0,42 ; intervalle de confiance à 95 % de -0,73 à -0,11 ; I² = 0 % ; 3 études ; 166 participants ; niveau de certitude modéré.
Les auteurs restent très incertains quant à l’effet du traitement pharmacologique sur les tests d’effort cardio-pulmonaire : différence moyenne standardisée -0,16 ; intervalle de confiance à 95 % de -0,46 à 0,13 ; I² = 0 % ; 3 études ; 181 participants ; niveau de certitude très faible.
Ils sont également très incertains quant à l’effet du traitement pharmacologique sur les effets indésirables liés au traitement : risque relatif 0,88 ; intervalle de confiance à 95 % de 0,63 à 1,22 ; I² = 0 % ; 2 études ; 104 participants ; niveau de certitude très faible. Seuls 104 participants étaient inclus dans cette analyse sur un total de 545 participants, ce qui limite fortement la portée de ce résultat.
La méta-analyse de six études a montré qu’il pourrait y avoir peu ou pas de différence entre les traitements pharmacologiques et le placebo concernant les effets indésirables apparus sous traitement conduisant à l’arrêt du médicament ou au décès : risque relatif 1,24 ; intervalle de confiance à 95 % de 0,44 à 3,48 ; I² = 0 % ; 6 études ; 313 participants ; niveau de certitude faible.
Le niveau de certitude des données allait de très faible à modéré. Les auteurs ont diminué le niveau de certitude de tous les critères de jugement d’un ou deux niveaux en raison d’un manque de précision, avec des diminutions supplémentaires liées à l’hétérogénéité des résultats — pour l’épaisseur du septum interventriculaire en diastole, les activités de la vie quotidienne et les tests d’effort cardio-pulmonaire — ainsi qu’à une suspicion de biais de publication pour ces mêmes critères et pour les effets indésirables.
Conclusions des auteurs :
Dans cette revue systématique Cochrane mise à jour, la méta-analyse des résultats portant sur les échelles d’évaluation de l’ataxie montre que les traitements pharmacologiques font probablement peu ou pas de différence par rapport au placebo après 12 mois de traitement. Compte tenu de ce résultat, l’amélioration probable observée de la dextérité des membres supérieurs était inattendue. Les effets indésirables apparus sous traitement conduisant à l’arrêt du médicament ou au décès pourraient ne pas être plus fréquents dans les groupes traités que dans les groupes placebo, car peu d’événements indésirables ont été détectés dans les groupes traités. Toutefois, les études n’ont peut-être pas permis de détecter tous les effets indésirables rares et graves.
[1] Un essai contrôlé randomisé sert à savoir, de la façon la plus fiable possible, si un traitement fonctionne mieux qu’un placebo ou qu’un traitement de référence. Le mot randomisé signifie que les participants sont répartis au hasard dans les groupes. Cela évite que les médecins ou les chercheurs choisissent eux-mêmes qui reçoit le traitement, ce qui pourrait fausser les résultats. Le mot contrôlé signifie qu’il existe un groupe de comparaison. Sans ce groupe, il serait difficile de savoir si une amélioration vient vraiment du médicament, de l’évolution naturelle de la maladie, de l’effet placebo ou d’autres facteurs.
[2] Le septum interventriculaire est la cloison musculaire qui sépare le ventricule droit et le ventricule gauche. La diastole correspond au moment où le cœur se relâche et se remplit de sang, entre deux contractions. Dans l’ataxie de Friedreich, le cœur peut être atteint, avec parfois un épaississement du muscle cardiaque, appelé cardiomyopathie hypertrophique. Mesurer cette l’ épaisseur du septum interventriculaire permet donc d’évaluer si un traitement a un effet sur l’atteinte cardiaque : par exemple, s’il réduit, stabilise ou n’améliore pas cet épaississement.
[3] Une méta-analyse est une méthode qui consiste à regrouper les résultats de plusieurs études portant sur la même question.
[4] La différence moyenne compare directement la moyenne des résultats entre deux groupes, par exemple : le groupe traité a diminué son score d’ataxie de 3 points, tandis que le groupe placebo l’a diminué de 1 point. La différence moyenne est donc de 2 points. La différence moyenne standardisée est utilisée quand plusieurs études mesurent le même phénomène avec des échelles différentes. Par exemple, une étude utilise une échelle d’ataxie sur 100 points, une autre sur 40 points. On transforme alors les résultats dans une unité commune pour pouvoir les comparer et les regrouper.
[5] Un critère continu est une mesure exprimée avec des chiffres sur une échelle : par exemple un score d’ataxie, une épaisseur cardiaque en millimètres, un poids, une tension artérielle.
[6] Un critère dichotomique est un résultat qui n’a que deux possibilités : oui/non, présent/absent, vivant/décédé, arrêt du traitement/pas d’arrêt du traitement. Le risque relatif compare la fréquence d’un événement entre deux groupes.
[7] Un intervalle de confiance à 95 % indique la marge d’incertitude autour d’un résultat.
[8] La méthode GRADE est une méthode utilisée pour dire à quel point on peut avoir confiance dans les résultats d’une étude ou d’une méta-analyse. Elle est notamment utilisée dans les revues Cochrane pour classer la certitude des preuves en quatre niveaux : élevée, modérée, faible ou très faible. En pratique, GRADE ne demande pas seulement : « le traitement semble-t-il marcher ? » Elle demande surtout : « Les données sont-elles assez solides pour croire ce résultat ? »
Dans cet article publié dans Lancet Neurology en mai 2026, les auteurs rapportent les résultats d’un essai de phase 2 évaluant l’efficacité d’un traitement combiné associant nicotinamide riboside et exercice physique dans l’ataxie de Friderich
Résumé de l’article traduit par l’équipe recherche.
(Des notes en bas de page visent à aider à la compréhension)
Contexte L’ataxie de Friedreich est une maladie rare, chronique, progressive et neurodégénérative, qui atteint plusieurs systèmes de l’organisme, notamment les systèmes neurologique, musculo-squelettique, cardiaque et endocrinien. Elle se caractérise aussi par une faible capacité cardio-pulmonaire. Nous avons testé l’effet de l’exercice physique et d’une supplémentation en précurseur du NAD⁺ par nicotinamide riboside, deux interventions ayant chacune montré des bénéfices dans des études animales et dans des études cliniques précoces, sur la capacité cardio-pulmonaire de personnes atteintes d’ataxie de Friedreich.
Méthodes
Il s’agissait d’un essai clinique de phase 2[1], ambulatoire[2], monocentrique[3], randomisé[4], avec un plan factoriel 2 × 2[5], d’une durée de 12 semaines. L’étude a été menée au Children’s Hospital of Philadelphia, à Philadelphie, aux États-Unis. Elle a recruté des personnes âgées de 10 à 40 ans, ayant une fraction d’éjection d’au moins 45 % et capables de faire de l’exercice.
Une séquence de randomisation générée par ordinateur a été élaborée par le statisticien de l’essai. La répartition aléatoire était stratifiée selon l’âge — moins de 18 ans versus 18 ans ou plus — dans l’un des quatre groupes suivants :
placebo sans exercice, avec contrôle attentionnel par appels téléphoniques hebdomadaires — appelé ensuite groupe placebo seul ;
nicotinamide riboside sans exercice, avec contrôle attentionnel — appelé ensuite groupe nicotinamide riboside seul ;
placebo avec exercice — appelé ensuite groupe exercice seul ;
nicotinamide riboside avec exercice — appelé ensuite traitement combiné.
Des programmes d’exercice individualisés ont été élaborés par le physiologiste de l’exercice. Ils comprenaient trois séances d’entraînement aérobie et deux séances de renforcement musculaire par semaine. Les séances étaient réalisées au domicile des participants et supervisées à distance, notamment par des appels téléphoniques du physiologiste.
La dose de nicotinamide riboside ou de placebo était adaptée au poids :
300 mg, soit 1 capsule, pour un poids de 24 kg à moins de 48 kg ;
600 mg, soit 2 capsules, pour un poids de 48 kg à moins de 72 kg ;
900 mg, soit 3 capsules, pour un poids supérieur à 72 kg.
Le critère principal était la variation du pic de VO₂[6], exprimé en L/min, mesuré lors d’un test d’effort cardio-pulmonaire après 12 semaines par rapport à la valeur initiale. L’effet du groupe de traitement a été évalué à l’aide d’un modèle statistique tenant compte de l’âge, qui était la variable de stratification, du sexe et du pic de VO₂ initial.
L’analyse de première étape comparait chaque traitement actif au groupe contrôle. L’analyse de deuxième étape, si le traitement combiné était efficace, comparait le traitement combiné à l’exercice seul. Le risque global d’erreur de type 1 [7]était maintenu à moins de 0,05. Les analyses ont été réalisées en intention de traiter[8]. Les événements indésirables ont été recueillis de manière systématique. L’essai est enregistré sur ClinicalTrials.gov sous le numéro NCT04192136 et il est terminé.
Résultats
Entre le 3 septembre 2020 et le 23 avril 2025, 74 personnes ont été incluses, dont 66 répondaient aux critères d’éligibilité et ont été réparties aléatoirement dans les quatre groupes de l’étude. Tous les participants ont terminé l’étude.
Parmi eux, 33 participants, soit 50 %, étaient des enfants âgés de 10 à 17 ans, et 33, soit 50 %, étaient des adultes âgés de 18 ans ou plus. Trente-sept participants, soit 56 %, étaient de sexe masculin, et 29, soit 44 %, de sexe féminin.
Les moyennes des moindres carrés[9] pour la variation du pic de VO₂, en L/min, étaient les suivantes :
-0,05 ; intervalle de confiance à 95 % de -0,16 à 0,06, chez les 17 participants du groupe contrôle ;
0,06 ; intervalle de confiance à 95 % de -0,05 à 0,17, chez les 17 participants du groupe nicotinamide riboside sans exercice ;
0,11 ; intervalle de confiance à 95 % de 0,00 à 0,22, chez les 16 participants du groupe placebo avec exercice ;
0,16 ; intervalle de confiance à 95 % de 0,05 à 0,27, chez les 16 participants du groupe nicotinamide riboside avec exercice.
Les différences entre les traitements actifs et le groupe contrôle étaient :
0,10 ; intervalle de confiance à 95 % de -0,05 à 0,26 ; p ajusté = 0,188, pour le nicotinamide riboside sans exercice ;
0,16 ; intervalle de confiance à 95 % de 0,00 à 0,31 ; p ajusté = 0,103, pour le placebo avec exercice ;
0,21 ; intervalle de confiance à 95 % de 0,05 à 0,36 ; p ajusté = 0,0299, pour l’association nicotinamide riboside et exercice[10].
Le traitement combiné n’était pas statistiquement différent de l’exercice seul : différence de -0,05 ; intervalle de confiance à 95 % de -0,10 à 0,21 ; p = 0,49.
Tous les événements indésirables étaient légers ou modérés. Ils comprenaient des symptômes gastro-intestinaux, des chutes, des infections des voies respiratoires supérieures et des éruptions cutanées.
Au moins un événement indésirable modéré d’intérêt dans ces catégories a été rapporté chez :
7 participants, soit 41 %, dans le groupe contrôle ;
6 participants, soit 35 %, dans le groupe nicotinamide riboside sans exercice ;
3 participants, soit 19 %, dans le groupe placebo avec exercice ;
4 participants, soit 25 %, dans le groupe nicotinamide riboside avec exercice.
Interprétation
L’association de nicotinamide riboside et d’exercice physique pendant 12 semaines était sûre et a augmenté la capacité cardio-pulmonaire chez des enfants et des adultes atteints d’ataxie de Friedreich.
Des études plus longues sont nécessaires pour établir si l’ajout du nicotinamide riboside à l’exercice physique pourrait être envisagé comme élément d’une stratégie thérapeutique globale à long terme.
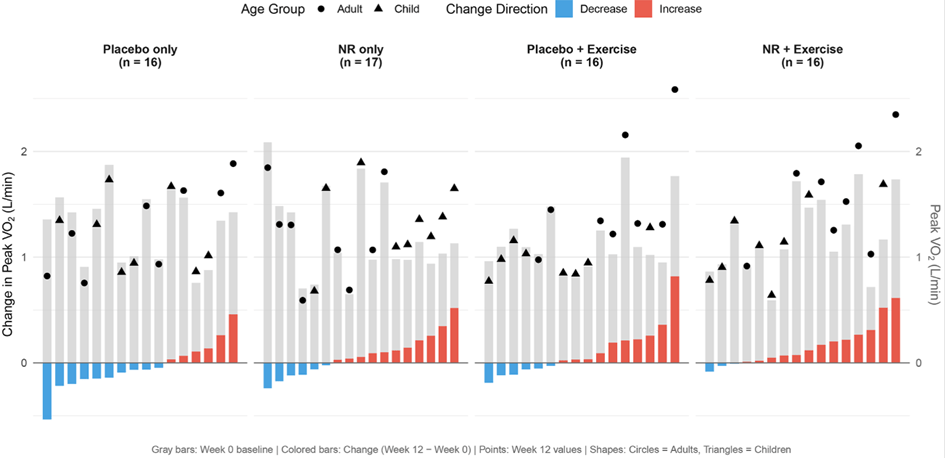
Figure 2. Pic de VO₂ (L/min), variation entre la valeur initiale et 12 semaines (diagramme en cascade).
Pour chaque participant, la variation du pic de VO₂ entre la semaine 0 et la semaine 12 est représentée, en L/min, selon le groupe de traitement. Les augmentations sont indiquées en rouge, et les diminutions en bleu. Les barres grises montrent les valeurs initiales à la semaine 0.
Les cercles représentent les adultes, âgés de 18 ans ou plus, et les triangles représentent les enfants, âgés de moins de 18 ans. Ils indiquent les valeurs à la semaine 12 ; là encore, les valeurs ayant augmenté par rapport au niveau initial sur les 12 semaines sont en rouge, et celles ayant diminué sont en bleu.
Le nombre de personnes incluses dans chaque analyse est indiqué par N. Dans le groupe placebo seul, une personne n’a pas réalisé le test d’effort cardio-pulmonaire à la semaine 12 en raison d’une blessure au pied ; elle n’a donc pas été incluse dans les résultats portant sur ce test.
[1] Un essai clinique de phase 2 est une étude réalisée chez des patients pour savoir si un traitement est prometteur et suffisamment sûr pour être étudié plus largement.
[2] Ambulatoire : les participants ne sont pas hospitalisés. Ils viennent pour les évaluations, mais vivent chez eux pendant l’étude.
[3] Monocentrique : l’étude est réalisée dans un seul centre médical, ici le Children’s Hospital of Philadelphia.
[4] Randomisé : les participants sont répartis au hasard dans les différents groupes, pour éviter de fausser les résultats.
[5] Plan factoriel 2 × 2 : l’étude teste deux interventions en même temps :
le nicotinamide riboside ou son placebo ;
l’exercice physique ou l’absence d’exercice.
Cela donne quatre groupes. Cette étude permet de comparer séparément l’effet du nicotinamide riboside, l’effet de l’exercice, et l’effet de leur association.
[6] Le pic de VO₂ est mesuré lors d’un test d’effort cardio-pulmonaire. Il indique la quantité maximale d’oxygène que la personne peut utiliser quand elle fait un exercice intense. La variation du pic de VO₂ correspond au changement de la capacité maximale du corps à utiliser l’oxygène pendant un effort.
[7] Le risque d’erreur de type 1 correspond au risque de conclure à tort qu’un traitement est efficace alors qu’en réalité il ne l’est pas. C’est comme dire : « il y a un effet du traitement », alors que cet effet est dû au hasard. Le risque global d’erreur de type 1 devient important quand une étude fait plusieurs comparaisons statistiques. Plus on multiplie les tests, plus on augmente le risque de trouver par hasard un résultat “significatif”.
[8] Les analyses réalisées en intention de traiter signifient que chaque participant est analysé dans le groupe où il a été tiré au sort au départ, même s’il n’a pas parfaitement suivi le traitement ou le programme prévu.
[9] La moyenne des moindres carrés est une moyenne ajustée statistiquement.
Elle ne correspond pas simplement à la moyenne brute observée dans un groupe. Elle tient compte de certains facteurs qui peuvent influencer le résultat ( par exemple ici l’âge, le sexe, et la valeur du pic de VO2 initial)
[10] Ce résultat est considéré comme statistiquement significatif, pour le traitement combiné versus placebo, car : Association nicotinamide riboside + exercice : différence = 0,21 ; intervalle de confiance à 95 % 0,05 à 0,36 ; p ajusté = 0,0299.
la valeur de p ajusté est inférieure à 0,05 ;
l’intervalle de confiance est entièrement au-dessus de 0 : il va de 0,05 à 0,36 ;
cela signifie que l’amélioration observée du pic de VO₂ est peu compatible avec le simple hasard statistique.
Gunther, K., Profeta, V., Keita, M. et al. Safety Monitoring of Omaveloxolone in Friedreich Ataxia: Results from One Year of Clinical Treatment. Neurol Ther14, 1105–1114 (2025). https://doi.org/10.1007/s40120-025-00749-3
Date de publication : Article publié le 30 avril 2025 par Gunther et al., dans la revue Neurology therapeutics
Traduction du résumé de l’article par le groupe Recherche de l’AFAF
Introduction
L’ataxie de Friedreich (AF), l’ataxie héréditaire la plus fréquente, se caractérise par une ataxie progressive de la démarche et des membres, une scoliose, une dysarthrie, une perte de la vue et une cardiomyopathie hypertrophique. L’âge d’apparition de la maladie se situe généralement dans les deux premières décennies de la vie, et les individus sont confinés au fauteuil roulant dans les 10 à 15 ans qui suivent l’apparition de la maladie; l’insuffisance cardiaque associée à la cardiomyopathie est la principale cause de mortalité. L’AF est généralement causée (96%) par des répétitions trinucléotidiques bialléliques de guanineadénine-adénine (GAA) dans l’intron 1 du gène FXN. Ces répétitions GAA expansées répriment l’expression de la FXN, ce qui entraîne une diminution des taux de la protéine frataxine. La frataxine pénètre dans les mitochondries, où elle facilite la biosynthèse des clusters fer-soufre (ISC) et la production d’énergie dans la cellule.
Alors que la thérapie définitive nécessite probablement de la frataxine, une variété d’approches visant à inverser le dysfonctionnement mitochondrial a fait l’objet d’essais cliniques, l’omaveloxolone étant approuvée par la FDA en 2023 . L’omaveloxolone stabilise le NRF2, un facteur de transcription qui contrôle la réponse endogène de la mitochondrie au stress oxydatif ; NRF2 est paradoxalement régulé à la baisse dans la l’AF. Dans les essais cliniques MOXIe menés auprès de patients atteints de AF âgés de 16 à 40 ans, l’omaveloxolone a montré une courbe de réponse de concentration avec un bénéfice maximal à 160 mg par jour, ce qui a conduit à un dosage de 150 mg par jour dans les études pivot. À cette dose, l’omaveloxolone a permis d’améliorer les scores mFARS, la mesure la plus couramment utilisée pour l’évaluation de la fonction neurologique dans l’AF, à 1 an, et les bénéfices ont persisté pendant une extension ouverte d’au moins 3 ans. Le médicament était également sûr et bien toléré dans l’étude MOXIe, mais sa posologie est compliquée par les interactions avec les inhibiteurs puissants du CYP4A3. Les inhibiteurs du CYP4A3 augmentent le métabolisme de l’omaveloxolone, ce qui nécessite une réduction de la posologie afin d’éviter une surmédication. Sur la base de ces données, l’omaveloxolone a été approuvée pour le traitement des patients atteints d’AF≥ 16 ans, sans aucune autre restriction. La surveillance recommandée du traitement par l’omaveloxolone comprend des taux annuels de BNP et de cholestérol ainsi qu’un test de la fonction hépatique au début du traitement et aux mois 1, 2, 3, 6 et 12 après le début du traitement. Nous rapportons ici la sécurité et la tolérabilité de l’omaveloxolone administrée en clinique pendant 1 an
MÉTHODES
Patients
Les procédures ont été approuvées par le l’Hopital de Philadelphie (CHOP), dans le cadre de l’étude de longue durée sur l’histoire naturelle de l’AF, FACOMS, et de son successeur (UNIFAI) (CHOP IRB #2609). Les sujets ont donné leur consentement éclairé dans le cadre de cette étude et ont été initialement identifiés comme étant ceux qui ont été suivis au CHOP dans le cadre de l’étude FACOMS. Les données ont été extraites de leurs visites d’étude et ont permis d’évaluer les antécédents médicaux, les tests de laboratoire et les événements indésirables.
Approbation éthique
L’étude a été approuvée par le CHOP IRB sous le nom d’étude 2609 et a été réalisée conformément à la déclaration d’Helsinki de 1964. Tous les sujets ont donné leur consentement éclairé pour participer à l’étude.
Collecte des données
Nous avons extrait la date de début, les valeurs de laboratoire de base et les interactions médicamenteuses pour chaque sujet. Les valeurs de laboratoire comprenaient les panels lipidiques (cholestérol total et LDL) et le peptide natriurétique cérébral (BNP). Lors des visites initiales et des tests de la fonction hépatique aux mois 1, 2, 3, 6 et 12 après le début de l’utilisation de l’omaveloxolone. En se concentrant sur l’alanine aminotransférase (ALT), l’aspartate aminotransférase (AST), l’albumine et la bilirubine totale. Pour les patients ayant déjà participé à l’essai clinique de l’omaveloxolone, seules les valeurs de laboratoire à 12 mois ont été extraites
Posologies
Tous les patients ont reçu la posologie recommandée de 150 mg/jour (trois comprimés de 50 mg), à l’exception de ceux qui prenaient des inhibiteurs chroniques du CYP3A4 (le plus souvent le diltiazem et le vérapamil). Les patients sous ces inhibiteurs ont reçu une dose de 100 mg/jour (deux comprimés) pour obtenir des concentrations sanguines présumées similaires [22]. Pour les sujets suivis cliniquement au CHOP, la dose d’omaveloxolone a été réduite en réponse à des transaminases élevées, comme suit : Les patients ont continué à recevoir la même dose si les valeurs de l’ALT étaient < 2×la LSN. Si la valeur de l’ALT était > 3×la limite supérieure de la normale (LSN), le patient a reçu pour instruction d’arrêter l’omaveloxolone pendant 2 semaines et de répéter les tests de laboratoire. Si les nouveaux tests montraient un taux d’ALT < 3 × la normale, le patient reprenait le traitement à 50 mg par jourde moins que la dose précédente. Une fois que le patient avait des taux d’ALT stables inférieurs à 2×ULN sur la base de tests en série (généralement tous les mois), les patients pouvaient augmenter leur dose de 50 mg (généralement tous les mois), les patients et revenir à la dose complète. Si le taux d’ALT était compris entre 2 et 2,9×ULN, les patients continuaient à recevoir la même dose, mais les valeurs de la fonction hépatique étaient réévaluées toutes les 2 semaines pour confirmer la stabilité des taux d’ALT. La limite supérieure de la normale (LSN) de l’ALT était en moyenne de 41,3 U/l, mais elle variait selon les caractéristiques démographiques du patient et le site d’évaluation. La valeur moyenne de chaque test est fournie à titre de référence. Pour les patients ayant subi une réduction de dose en raison des valeurs de l’ALT ou d’autres effets secondaires, le dosage a été lentement ramené au schéma posologique initial si les valeurs des transaminases restaient < 3×normales
Résultats
Deux cent quatre-vingt-douze patients ont été identifiés pour l’étude ; 236 ont eu une prescription d’omaveloxolone par un membre de la division de neurologie du CHOP (et ont participé au programme FACOMS/UNIFAI au CHOP) tandis que 56 autres patients ont eu recours à des prescripteurs externes (en obtenant d’autres soins ou en participant au FACOMS/UNIFAI au CHOP). Au CHOP, l’âge moyen au début du traitement était de 31,8 ans au CHOP et de 28,8 ans pour les autres. La durée moyenne de la maladie était de 17,8. La durée moyenne de la maladie était de 17,8 ans (CHOP) et de 18,9 ans (non-CHOP). L’âge moyen d’apparition de la maladie était de 13,9 (CHOP) et 10,3 (non-CHOP). La longueur moyenne des répétitions GAA1 était de 631,7 (CHOP) et de 720 (non-CHOP). Au CHOP, 44,4 % des patients étaient des hommes et 55,5 % des femmes. Les patients prescrits en dehors du CHOP étaient à 50 % des hommes et à 50 % des femmes. Quarante personnes avaient participé aux essais MOXIe, dont 33 sont passées directement de l’étude à la commercialisation. Les sept autres patients ont participé à MOXIe mais ont mis fin à leur participation avant de commencer à prendre le médicament commercial. L’analyse s’est concentrée sur les personnes pour lesquelles l’omaveloxolone a été prescrite au CHOP car des informations plus détaillées étaient disponibles. Sur les 236 patients auxquels l’omaveloxolone a été prescrite au CHOP, 207 ont continué à prendre le médicament, la plupart d’entre eux (88,4 %) l’ayant pris pendant plus d’un an. Cinq patients ont eu un refus de prise en charge et 21 patients ont arrêté l’omaveloxolone l’omaveloxolone (tableau 2). Les patients ont principalement abandonné l’omaveloxolone en raison d’effets secondaires signalés ou ont été perdus de vue. Parmi les patients traités au CHOP, 22 ont obtenu des médicaments par le biais d’un programme d’aide aux patients, tandis que l’assurance du patient couvrait l’omaveloxolone chez 214 personnes
Posologie
La majorité des patients traités au CHOP ont atteint la dose maximale de 150 mg par jour (trois comprimés de 50 mg par jour) ; 87,9 % des patients ont une dose maximale idéale de 150 mg/jour, et 12,1 % ont une dose maximale de 100 mg (2 pilules) par jour en raison d’interactions avec d’autres médicaments. Quatre-vingt-cinq pour cent (176 patients) traités au CHOP atteignent 150 mg par jour, 14,0 % (29 patients) prenait 100 mg par jour, et 12,1 % prenait 100 mg par jour et 1,0 % (2 patients) prenait 50 mg (une pilule) par jour. Sept patients patients prennent actuellement (au moins 12 mois après l’instauration du traitement) moins que leur dose idéale mais cherchent à atteindre la dose idéale maximale
Événements indésirables
Dans la cohorte, 63 sujets ont rapporté des événements indésirables significatifs, correspondant le plus souvent à des effets secondaires, correspondant le plus souvent à ceux observés dans l’essai clinique (troubles gastro-intestinaux, céphalées), mais quelques personnes ont interrompu leur traitement en raison de ces événements. Certains patients ont présenté plusieurs effets indésirables, qui ont presque tous disparus au cours des 3 premiers mois (tableau 3). Trois personnes ont signalé des éruptions cutanées, en corrélation étroite avec l’administration d’omaveloxolone pour l’un d’entre eux. Pour deux de ces patients, un consultant en dermatologie a estimé que l’éruption n’était pas liée après une biopsie de la peau. Parmi les patients présentant une éruption cutanée, deux ont toléré le traitement, deux d’entre eux ont toléré le médicament avec une réaction cutanée minime lors de la ré-introduction du traitement, tandis que le troisième a eu une récidive et a arrêté le traitement définitivement. Aucun effet secondaire n’a été observé chez les personnes qui ont quitté l’étude MOXIe.
Surveillance de la fonction hépatique
Comme prévu, des élévations des transaminases (toute valeur supérieure à la limite supérieure de la normale) étaient constatées chez de nombreux sujets, 56,6 % des patients ayant eu une élévation des transaminases à un moment ou à un autre au cours de la période de 12 mois (p<0,0001).
Au bout de 12 mois, 16,6 % des patients présentaient une élévation des transaminases (p<0,0001), mais aucune n’était supérieure à 3 fois la limite supérieure de la normale (Fig. 1A, B). L’omaveloxolone n’a pas modifié les taux d’albumine ou de bilirubine totale chez un grand nombre de patients, quel que soit le moment. Un patient a présenté une baisse significative d’albumine en raison d’une pneumonie non liée à l’omaveloxolone. Deux autres élévations significatives de bilirubine ont été expliquées par des comorbidités non liées (syndrome de Gilbert et atrésie des voies biliaires). Dans ces situations, de majoration de l’albumine ou la bilirubine , on a estimé qu’elles étaient liées à des comorbidités plutôt qu’à l’AF ou à l’omaveloxone et n’étaient pas en corrélation avec les variations des transaminases. (Fig. 1C, D).
Aucune anomalie des tests de la fonction hépatique n’a été chez ceux qui avaient été transférés directement de l’essai MOXIe. Vingt-six patients ont interrompu le traitement en raison d’une élévation des transaminases, et un patient a interrompu temporairement le traitement en raison d’effets secondaires. Parmi les 27 patients qui ont interrompu leur traitement, 23 sont revenus à leur posologie cible. Les quatre patients restants ont continué à augmenter leur posologie pour atteindre leur objectif. Quatre patients ont décidé d’arrêter définitivement le traitement, et un seul patient (avec une éruption cutanée) n’a pas pu reprendre un traitement modifié ou complet.
La cohorte comprenait trois personnes qui avaient été retirés du traitement actif dans l’étude MOXIe en raison d’une élévation persistante des transaminases >5×normale. Ces personnes ont commencé à recevoir de l’omaveloxolone ; avec une surveillance étroite de la fonction hépatique. Tous les individus ont à nouveau présenté une augmentation spectaculaire des transaminases, ce qui a nécessité une réduction de la dose. Cependant, ils ont pu reprendre le traitement à l’omaveloxolone et, après 1 an, deux d’entre eux ont atteint leur posologie cible (Fig. 2)
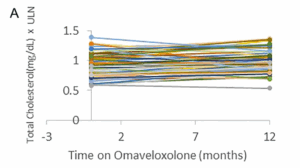
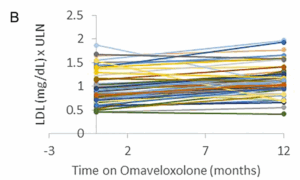
Autres valeurs de laboratoire
Les taux de cholestérol total et de LDL étaient légèrement élevés en moyenne après 12 mois de traitement. La limite supérieure de la normale (LSN) pour le cholestérol total était de 197,0 mg/dl et la limite supérieure moyenne pour les LDL était de 108,9 mg/dl. En moyenne, on a observé une augmentation de 0,07 (±0,16) point dans les taux de cholestérol total au-dessus de la LSN et une augmentation de 0,24 (±0,16) du taux de LDL au-dessus de la LSN (Fig. 3). De même, le BNP n’a pas connu de changement significatif au cours de la période d’un an. La LSN pour le BNP est de 100 pg/ml. Trois patients ayant des antécédents de dysfonctionnement cardiaque (fraction d’éjection basse) ont eu une légère augmentation du BNP, et un patient a vu son taux baisser, ce qui suggère que l’omaveloxolone n’a pas d’effet constant sur le BNP (Fig. 3).
DISCUSSION
L’utilisation clinique de l’omaveloxolone au cours des 19 premiers mois suivant sa mise sur le marché récapitule la plupart des résultats de l’essai MOXIe en matière de sécurité. Jusqu’à présent, peu d’effets indésirables majeurs sont survenus avec le traitement à l’omaveloxolone, que ce soit dans l’étude MOXIe ou dans l’utilisation clinique décrite ici. Ces résultats justifient son utilisation continue et suggèrent que l’étude MOXIe, en tant qu’essai clinique représentatif, reflète les effets de l’omaveloxolone dans des populations plus larges atteintes d’AF.
Néanmoins, divers problèmes peuvent survenir avec l’omaveloxolone. Si l’accès à l’omaveloxolone a été difficile pour certains sujets, la grande majorité d’entre eux ont fini par y avoir accès. De manière surprenante, alors que tous les patients ont reçu l’instruction d’obtenir des données de laboratoire avant de commencer à prendre de l’omaveloxolone, seuls 57 % d’entre eux avaient des transaminases de base. Ce faible taux pourrait refléter le fait que la population de patients n’est pas familiarisée avec la surveillance des médicaments. mais il est également probable qu’il résulte du délai entre l’approbation (28 février 2023) et la mise sur le marché (7 juillet 2023) du médicament. Une fois le traitement commencé, les anomalies de laboratoire étaient modestes. Les anomalies de la fonction hépatique se sont limitées aux transaminases et se sont résorbées avec le temps. en association avec l’interruption ou la réduction de la dose. Cela correspond le mieux à un effet métabolique du foie plutôt qu’une toxicité typique. Malgré les effets du composé apparenté bardoxolone, aucun effet n’a été noté sur les valeurs de BNP, et les changements de cholestérol, bien que présents, étaient modestes [23]. Les effets secondaires étaient également modestes et transitoires. En outre, à l’exception d’une personne ayant présenté une éruption cutanée importante, aucun nouvel effet indésirable n’est apparu.
CONCLUSION
Dans l’ensemble, les patients ont bien répondu au traitement commercial par l’omaveloxolone. Les élévations des transaminases étaient attendues et se sont stabilisées avec le temps ou après modification de la dose. Les effets indésirables ont été modestes et similaires à ceux rapportés dans l’étude MOXIe. D’autres études devraient porter sur les bénéfices rapportés et les changements dans l‘échelle mFARS après 1 an de traitement. D’autres investigations devraient également examiner les changements dans les maladies cardiaques
Références : Munoz-Zuluaga C, Gertz M, Yost-Bido M, Greco A, Gorman N, Chen A, Kooner V, Rosenberg JB, De BP, Kaminsky SM, Bborczuk A, Ricart Arbona R, Martin HR, Monette S, Khanna R, Barth JA, Crystal RG, Sondhi D. Hum Gene Ther. 2023 May 11.
Pour offrir les meilleures expériences, nous utilisons des technologies telles que les cookies pour stocker et/ou accéder aux informations des appareils. Le fait de consentir à ces technologies nous permettra de traiter des données telles que le comportement de navigation ou les ID uniques sur ce site. Le fait de ne pas consentir ou de retirer son consentement peut avoir un effet négatif sur certaines caractéristiques et fonctions.
Fonctionnel
Toujours activé
Le stockage ou l’accès technique est strictement nécessaire dans la finalité d’intérêt légitime de permettre l’utilisation d’un service spécifique explicitement demandé par l’abonné ou l’utilisateur, ou dans le seul but d’effectuer la transmission d’une communication sur un réseau de communications électroniques.
Préférences
Le stockage ou l’accès technique est nécessaire dans la finalité d’intérêt légitime de stocker des préférences qui ne sont pas demandées par l’abonné ou l’utilisateur.
Statistiques
Le stockage ou l’accès technique qui est utilisé exclusivement à des fins statistiques.Le stockage ou l’accès technique qui est utilisé exclusivement dans des finalités statistiques anonymes. En l’absence d’une assignation à comparaître, d’une conformité volontaire de la part de votre fournisseur d’accès à internet ou d’enregistrements supplémentaires provenant d’une tierce partie, les informations stockées ou extraites à cette seule fin ne peuvent généralement pas être utilisées pour vous identifier.
Marketing
Le stockage ou l’accès technique est nécessaire pour créer des profils d’utilisateurs afin d’envoyer des publicités, ou pour suivre l’utilisateur sur un site web ou sur plusieurs sites web ayant des finalités marketing similaires.