Gunther, K., Profeta, V., Keita, M. et al. Safety Monitoring of Omaveloxolone in Friedreich Ataxia: Results from One Year of Clinical Treatment. Neurol Ther 14, 1105–1114 (2025). https://doi.org/10.1007/s40120-025-00749-3
Date de publication : Article publié le 30 avril 2025 par Gunther et al., dans la revue Neurology therapeutics
Article : Safety Monitoring of Omaveloxolone in Friedreich Ataxia: Results from One Year of Clinical Treatment
Traduction du résumé de l’article par le groupe Recherche de l’AFAF
Introduction
L’ataxie de Friedreich (AF), l’ataxie héréditaire la plus fréquente, se caractérise par une ataxie progressive de la démarche et des membres, une scoliose, une dysarthrie, une perte de la vue et une cardiomyopathie hypertrophique. L’âge d’apparition de la maladie se situe généralement dans les deux premières décennies de la vie, et les individus sont confinés au fauteuil roulant dans les 10 à 15 ans qui suivent l’apparition de la maladie; l’insuffisance cardiaque associée à la cardiomyopathie est la principale cause de mortalité. L’AF est généralement causée (96%) par des répétitions trinucléotidiques bialléliques de guanineadénine-adénine (GAA) dans l’intron 1 du gène FXN. Ces répétitions GAA expansées répriment l’expression de la FXN, ce qui entraîne une diminution des taux de la protéine frataxine. La frataxine pénètre dans les mitochondries, où elle facilite la biosynthèse des clusters fer-soufre (ISC) et la production d’énergie dans la cellule.
Alors que la thérapie définitive nécessite probablement de la frataxine, une variété d’approches visant à inverser le dysfonctionnement mitochondrial a fait l’objet d’essais cliniques, l’omaveloxolone étant approuvée par la FDA en 2023 . L’omaveloxolone stabilise le NRF2, un facteur de transcription qui contrôle la réponse endogène de la mitochondrie au stress oxydatif ; NRF2 est paradoxalement régulé à la baisse dans la l’AF. Dans les essais cliniques MOXIe menés auprès de patients atteints de AF âgés de 16 à 40 ans, l’omaveloxolone a montré une courbe de réponse de concentration avec un bénéfice maximal à 160 mg par jour, ce qui a conduit à un dosage de 150 mg par jour dans les études pivot. À cette dose, l’omaveloxolone a permis d’améliorer les scores mFARS, la mesure la plus couramment utilisée pour l’évaluation de la fonction neurologique dans l’AF, à 1 an, et les bénéfices ont persisté pendant une extension ouverte d’au moins 3 ans. Le médicament était également sûr et bien toléré dans l’étude MOXIe, mais sa posologie est compliquée par les interactions avec les inhibiteurs puissants du CYP4A3. Les inhibiteurs du CYP4A3 augmentent le métabolisme de l’omaveloxolone, ce qui nécessite une réduction de la posologie afin d’éviter une surmédication. Sur la base de ces données, l’omaveloxolone a été approuvée pour le traitement des patients atteints d’AF≥ 16 ans, sans aucune autre restriction. La surveillance recommandée du traitement par l’omaveloxolone comprend des taux annuels de BNP et de cholestérol ainsi qu’un test de la fonction hépatique au début du traitement et aux mois 1, 2, 3, 6 et 12 après le début du traitement. Nous rapportons ici la sécurité et la tolérabilité de l’omaveloxolone administrée en clinique pendant 1 an
MÉTHODES
Patients
Les procédures ont été approuvées par le l’Hopital de Philadelphie (CHOP), dans le cadre de l’étude de longue durée sur l’histoire naturelle de l’AF, FACOMS, et de son successeur (UNIFAI) (CHOP IRB #2609). Les sujets ont donné leur consentement éclairé dans le cadre de cette étude et ont été initialement identifiés comme étant ceux qui ont été suivis au CHOP dans le cadre de l’étude FACOMS. Les données ont été extraites de leurs visites d’étude et ont permis d’évaluer les antécédents médicaux, les tests de laboratoire et les événements indésirables.
Approbation éthique
L’étude a été approuvée par le CHOP IRB sous le nom d’étude 2609 et a été réalisée conformément à la déclaration d’Helsinki de 1964. Tous les sujets ont donné leur consentement éclairé pour participer à l’étude.
Collecte des données
Nous avons extrait la date de début, les valeurs de laboratoire de base et les interactions médicamenteuses pour chaque sujet. Les valeurs de laboratoire comprenaient les panels lipidiques (cholestérol total et LDL) et le peptide natriurétique cérébral (BNP). Lors des visites initiales et des tests de la fonction hépatique aux mois 1, 2, 3, 6 et 12 après le début de l’utilisation de l’omaveloxolone. En se concentrant sur l’alanine aminotransférase (ALT), l’aspartate aminotransférase (AST), l’albumine et la bilirubine totale. Pour les patients ayant déjà participé à l’essai clinique de l’omaveloxolone, seules les valeurs de laboratoire à 12 mois ont été extraites
Posologies
Tous les patients ont reçu la posologie recommandée de 150 mg/jour (trois comprimés de 50 mg), à l’exception de ceux qui prenaient des inhibiteurs chroniques du CYP3A4 (le plus souvent le diltiazem et le vérapamil). Les patients sous ces inhibiteurs ont reçu une dose de 100 mg/jour (deux comprimés) pour obtenir des concentrations sanguines présumées similaires [22]. Pour les sujets suivis cliniquement au CHOP, la dose d’omaveloxolone a été réduite en réponse à des transaminases élevées, comme suit : Les patients ont continué à recevoir la même dose si les valeurs de l’ALT étaient < 2×la LSN. Si la valeur de l’ALT était > 3×la limite supérieure de la normale (LSN), le patient a reçu pour instruction d’arrêter l’omaveloxolone pendant 2 semaines et de répéter les tests de laboratoire. Si les nouveaux tests montraient un taux d’ALT < 3 × la normale, le patient reprenait le traitement à 50 mg par jourde moins que la dose précédente. Une fois que le patient avait des taux d’ALT stables inférieurs à 2×ULN sur la base de tests en série (généralement tous les mois), les patients pouvaient augmenter leur dose de 50 mg (généralement tous les mois), les patients et revenir à la dose complète. Si le taux d’ALT était compris entre 2 et 2,9×ULN, les patients continuaient à recevoir la même dose, mais les valeurs de la fonction hépatique étaient réévaluées toutes les 2 semaines pour confirmer la stabilité des taux d’ALT. La limite supérieure de la normale (LSN) de l’ALT était en moyenne de 41,3 U/l, mais elle variait selon les caractéristiques démographiques du patient et le site d’évaluation. La valeur moyenne de chaque test est fournie à titre de référence. Pour les patients ayant subi une réduction de dose en raison des valeurs de l’ALT ou d’autres effets secondaires, le dosage a été lentement ramené au schéma posologique initial si les valeurs des transaminases restaient < 3×normales
Résultats
Deux cent quatre-vingt-douze patients ont été identifiés pour l’étude ; 236 ont eu une prescription d’omaveloxolone par un membre de la division de neurologie du CHOP (et ont participé au programme FACOMS/UNIFAI au CHOP) tandis que 56 autres patients ont eu recours à des prescripteurs externes (en obtenant d’autres soins ou en participant au FACOMS/UNIFAI au CHOP). Au CHOP, l’âge moyen au début du traitement était de 31,8 ans au CHOP et de 28,8 ans pour les autres. La durée moyenne de la maladie était de 17,8. La durée moyenne de la maladie était de 17,8 ans (CHOP) et de 18,9 ans (non-CHOP). L’âge moyen d’apparition de la maladie était de 13,9 (CHOP) et 10,3 (non-CHOP). La longueur moyenne des répétitions GAA1 était de 631,7 (CHOP) et de 720 (non-CHOP). Au CHOP, 44,4 % des patients étaient des hommes et 55,5 % des femmes. Les patients prescrits en dehors du CHOP étaient à 50 % des hommes et à 50 % des femmes. Quarante personnes avaient participé aux essais MOXIe, dont 33 sont passées directement de l’étude à la commercialisation. Les sept autres patients ont participé à MOXIe mais ont mis fin à leur participation avant de commencer à prendre le médicament commercial. L’analyse s’est concentrée sur les personnes pour lesquelles l’omaveloxolone a été prescrite au CHOP car des informations plus détaillées étaient disponibles. Sur les 236 patients auxquels l’omaveloxolone a été prescrite au CHOP, 207 ont continué à prendre le médicament, la plupart d’entre eux (88,4 %) l’ayant pris pendant plus d’un an. Cinq patients ont eu un refus de prise en charge et 21 patients ont arrêté l’omaveloxolone l’omaveloxolone (tableau 2). Les patients ont principalement abandonné l’omaveloxolone en raison d’effets secondaires signalés ou ont été perdus de vue. Parmi les patients traités au CHOP, 22 ont obtenu des médicaments par le biais d’un programme d’aide aux patients, tandis que l’assurance du patient couvrait l’omaveloxolone chez 214 personnes
Posologie
La majorité des patients traités au CHOP ont atteint la dose maximale de 150 mg par jour (trois comprimés de 50 mg par jour) ; 87,9 % des patients ont une dose maximale idéale de 150 mg/jour, et 12,1 % ont une dose maximale de 100 mg (2 pilules) par jour en raison d’interactions avec d’autres médicaments. Quatre-vingt-cinq pour cent (176 patients) traités au CHOP atteignent 150 mg par jour, 14,0 % (29 patients) prenait 100 mg par jour, et 12,1 % prenait 100 mg par jour et 1,0 % (2 patients) prenait 50 mg (une pilule) par jour. Sept patients patients prennent actuellement (au moins 12 mois après l’instauration du traitement) moins que leur dose idéale mais cherchent à atteindre la dose idéale maximale
Événements indésirables
Dans la cohorte, 63 sujets ont rapporté des événements indésirables significatifs, correspondant le plus souvent à des effets secondaires, correspondant le plus souvent à ceux observés dans l’essai clinique (troubles gastro-intestinaux, céphalées), mais quelques personnes ont interrompu leur traitement en raison de ces événements. Certains patients ont présenté plusieurs effets indésirables, qui ont presque tous disparus au cours des 3 premiers mois (tableau 3). Trois personnes ont signalé des éruptions cutanées, en corrélation étroite avec l’administration d’omaveloxolone pour l’un d’entre eux. Pour deux de ces patients, un consultant en dermatologie a estimé que l’éruption n’était pas liée après une biopsie de la peau. Parmi les patients présentant une éruption cutanée, deux ont toléré le traitement, deux d’entre eux ont toléré le médicament avec une réaction cutanée minime lors de la ré-introduction du traitement, tandis que le troisième a eu une récidive et a arrêté le traitement définitivement. Aucun effet secondaire n’a été observé chez les personnes qui ont quitté l’étude MOXIe.
Surveillance de la fonction hépatique
Comme prévu, des élévations des transaminases (toute valeur supérieure à la limite supérieure de la normale) étaient constatées chez de nombreux sujets, 56,6 % des patients ayant eu une élévation des transaminases à un moment ou à un autre au cours de la période de 12 mois (p<0,0001).
Au bout de 12 mois, 16,6 % des patients présentaient une élévation des transaminases (p<0,0001), mais aucune n’était supérieure à 3 fois la limite supérieure de la normale (Fig. 1A, B). L’omaveloxolone n’a pas modifié les taux d’albumine ou de bilirubine totale chez un grand nombre de patients, quel que soit le moment. Un patient a présenté une baisse significative d’albumine en raison d’une pneumonie non liée à l’omaveloxolone. Deux autres élévations significatives de bilirubine ont été expliquées par des comorbidités non liées (syndrome de Gilbert et atrésie des voies biliaires). Dans ces situations, de majoration de l’albumine ou la bilirubine , on a estimé qu’elles étaient liées à des comorbidités plutôt qu’à l’AF ou à l’omaveloxone et n’étaient pas en corrélation avec les variations des transaminases. (Fig. 1C, D).
Aucune anomalie des tests de la fonction hépatique n’a été chez ceux qui avaient été transférés directement de l’essai MOXIe. Vingt-six patients ont interrompu le traitement en raison d’une élévation des transaminases, et un patient a interrompu temporairement le traitement en raison d’effets secondaires. Parmi les 27 patients qui ont interrompu leur traitement, 23 sont revenus à leur posologie cible. Les quatre patients restants ont continué à augmenter leur posologie pour atteindre leur objectif. Quatre patients ont décidé d’arrêter définitivement le traitement, et un seul patient (avec une éruption cutanée) n’a pas pu reprendre un traitement modifié ou complet.
La cohorte comprenait trois personnes qui avaient été retirés du traitement actif dans l’étude MOXIe en raison d’une élévation persistante des transaminases >5×normale. Ces personnes ont commencé à recevoir de l’omaveloxolone ; avec une surveillance étroite de la fonction hépatique. Tous les individus ont à nouveau présenté une augmentation spectaculaire des transaminases, ce qui a nécessité une réduction de la dose. Cependant, ils ont pu reprendre le traitement à l’omaveloxolone et, après 1 an, deux d’entre eux ont atteint leur posologie cible (Fig. 2)
Autres valeurs de laboratoire
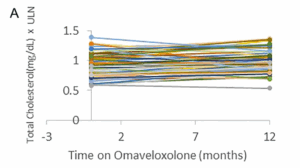
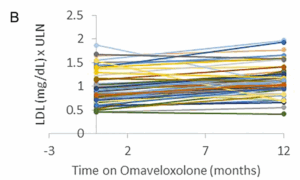
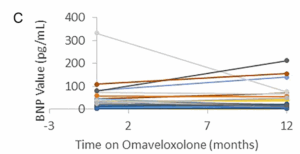
Les taux de cholestérol total et de LDL étaient légèrement élevés en moyenne après 12 mois de traitement. La limite supérieure de la normale (LSN) pour le cholestérol total était de 197,0 mg/dl et la limite supérieure moyenne pour les LDL était de 108,9 mg/dl. En moyenne, on a observé une augmentation de 0,07 (±0,16) point dans les taux de cholestérol total au-dessus de la LSN et une augmentation de 0,24 (±0,16) du taux de LDL au-dessus de la LSN (Fig. 3). De même, le BNP n’a pas connu de changement significatif au cours de la période d’un an. La LSN pour le BNP est de 100 pg/ml. Trois patients ayant des antécédents de dysfonctionnement cardiaque (fraction d’éjection basse) ont eu une légère augmentation du BNP, et un patient a vu son taux baisser, ce qui suggère que l’omaveloxolone n’a pas d’effet constant sur le BNP (Fig. 3).
DISCUSSION
L’utilisation clinique de l’omaveloxolone au cours des 19 premiers mois suivant sa mise sur le marché récapitule la plupart des résultats de l’essai MOXIe en matière de sécurité. Jusqu’à présent, peu d’effets indésirables majeurs sont survenus avec le traitement à l’omaveloxolone, que ce soit dans l’étude MOXIe ou dans l’utilisation clinique décrite ici. Ces résultats justifient son utilisation continue et suggèrent que l’étude MOXIe, en tant qu’essai clinique représentatif, reflète les effets de l’omaveloxolone dans des populations plus larges atteintes d’AF.
Néanmoins, divers problèmes peuvent survenir avec l’omaveloxolone. Si l’accès à l’omaveloxolone a été difficile pour certains sujets, la grande majorité d’entre eux ont fini par y avoir accès. De manière surprenante, alors que tous les patients ont reçu l’instruction d’obtenir des données de laboratoire avant de commencer à prendre de l’omaveloxolone, seuls 57 % d’entre eux avaient des transaminases de base. Ce faible taux pourrait refléter le fait que la population de patients n’est pas familiarisée avec la surveillance des médicaments. mais il est également probable qu’il résulte du délai entre l’approbation (28 février 2023) et la mise sur le marché (7 juillet 2023) du médicament. Une fois le traitement commencé, les anomalies de laboratoire étaient modestes. Les anomalies de la fonction hépatique se sont limitées aux transaminases et se sont résorbées avec le temps. en association avec l’interruption ou la réduction de la dose. Cela correspond le mieux à un effet métabolique du foie plutôt qu’une toxicité typique. Malgré les effets du composé apparenté bardoxolone, aucun effet n’a été noté sur les valeurs de BNP, et les changements de cholestérol, bien que présents, étaient modestes [23]. Les effets secondaires étaient également modestes et transitoires. En outre, à l’exception d’une personne ayant présenté une éruption cutanée importante, aucun nouvel effet indésirable n’est apparu.
CONCLUSION
Dans l’ensemble, les patients ont bien répondu au traitement commercial par l’omaveloxolone. Les élévations des transaminases étaient attendues et se sont stabilisées avec le temps ou après modification de la dose. Les effets indésirables ont été modestes et similaires à ceux rapportés dans l’étude MOXIe. D’autres études devraient porter sur les bénéfices rapportés et les changements dans l‘échelle mFARS après 1 an de traitement. D’autres investigations devraient également examiner les changements dans les maladies cardiaques